

Jernigan NL, Walker BR, Resta TC
Abstract:
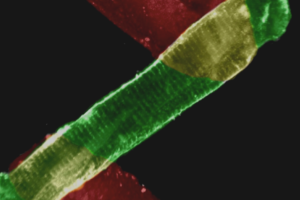
Recent evidence supports a prominent role for Rho kinase (ROK)-mediated pulmonary vasoconstriction in the development and maintenance of chronic hypoxia (CH)-induced pulmonary hypertension. Endothelin (ET)-1 contributes to the pulmonary hypertensive response to CH and recent studies by our laboratory and others indicate that pulmonary vascular reactivity following CH is largely independent of changes in vascular smooth muscle (VSM) intracellular free calcium concentration ([Ca2+]i). In addition, CH increases generation of reactive oxygen species (ROS) in pulmonary arteries, which may underlie the shift towards ROK-dependent Ca2+ sensitization. Therefore we hypothesized that ROS-mediated RhoA/ROK signaling mediates ET-1-induced Ca2+ sensitization in pulmonary VSM following CH. To test this hypothesis we determined the effect of pharmacological inhibitors of ROK, myosin light chain kinase (MLCK), tyrosine kinase (TK), and PKC on ET-1-induced vasoconstriction in endothelium-denuded, Ca2+-permeabilized small pulmonary arteries from control and CH (4 wk at 0.5 atm) rats. Further experiments examined ET-1-mediated, ROK-dependent phosphorylation of the regulatory subunit of myosin light chain phosphatase (MLCP), MYPT1. Finally, we measured ET-1-induced ROS generation in dihydroethidium (DHE)-loaded small pulmonary arteries and investigated the role of ROS in mediating ET-1-induced, RhoA/ROK-dependent Ca2+-sensitization using the superoxide anion scavenger, tiron. We found that CH increases ET-1-induced Ca2+-sensitization that is sensitive to inhibition of ROK and MLCK, but not PKC or TK, and correlates with ROK-dependent MYPT1Thr696 phosphorylation. Furthermore, tiron inhibited basal and ET-1-stimulated ROS generation, RhoA activation, and VSM Ca2+-sensitization following CH. We conclude that CH augments ET-1-induced Ca2+-sensitization through ROS-dependent activation of RhoA/ROK signaling in pulmonary VSM.